В панель Синдром Жубер входит 36 генов, также включена оценка некодирующих вариантов, обнаружение делеций и дупликаций.
Идеально подходит для пациентов с клиническим подозрением на синдром Жубера.
Гены на этой панели включены в панель Цилиопатии и панель Дистрофии сетчатки.
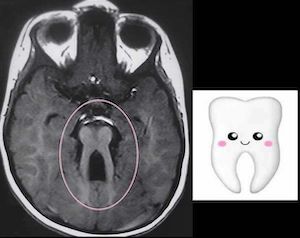 Классический синдром Жубера (JS) характеризуется тремя основными признаками: отличительной аномалией развития мозжечка и ствола мозга, так называемой признаком коренного зуба (MTS), гипотонией и задержкой развития. MTS, видимый на магнитно-резонансной томографии (МРТ), является результатом гипоплазии червя мозжечка и аномалий развития среднего и заднего мозга. Дополнительные признаки, связанные с синдромом Жубера, включают аномалии сетчатки, нистагм, атаксию, полидактилию, фиброз печени и заболевание почек. Оценки распространенности варьируются от 1:80 000 до 1:100 000, но это может быть заниженной оценкой. В целом, около 50% людей с синдромом Жубера и связанными с ним расстройствами имеют патогенные варианты, идентифицированные в одном из идентифицированных генов. Дефектная биология ресничек лежит в основе JS, и она разделяет существенный аллелизм с другими цилиопатиями.
Классический синдром Жубера (JS) характеризуется тремя основными признаками: отличительной аномалией развития мозжечка и ствола мозга, так называемой признаком коренного зуба (MTS), гипотонией и задержкой развития. MTS, видимый на магнитно-резонансной томографии (МРТ), является результатом гипоплазии червя мозжечка и аномалий развития среднего и заднего мозга. Дополнительные признаки, связанные с синдромом Жубера, включают аномалии сетчатки, нистагм, атаксию, полидактилию, фиброз печени и заболевание почек. Оценки распространенности варьируются от 1:80 000 до 1:100 000, но это может быть заниженной оценкой. В целом, около 50% людей с синдромом Жубера и связанными с ним расстройствами имеют патогенные варианты, идентифицированные в одном из идентифицированных генов. Дефектная биология ресничек лежит в основе JS, и она разделяет существенный аллелизм с другими цилиопатиями.
Гены, которые входят в панель:
| Ген |
Фенотип |
Наслед.* |
ClinVar** |
HGMD** |
|
AHI1
|
синдром Жубера |
AR |
62 |
93 |
|
ARL13B
|
синдром Жубера |
AR |
11 |
10 |
|
ARMC9
|
Синдром Жубера 30 |
AR |
12 |
11 |
|
B9D1
|
синдром Меккеля |
AR |
7 |
10 |
|
B9D2
|
синдром Меккеля |
AR |
8 |
4 |
|
C21ORF2
|
Дистрофия сетчатки с макулярной стафиломой или без нее (RDMS), спондилометафизарная дисплазия, аксиальная (SMDAX) |
AR |
13 |
22 |
|
C5ORF42
|
Орофациодигитальный синдром VI, синдром Жубера 17 |
AR |
97 |
103 |
|
CC2D2A
|
синдром COACH, синдром Жубера, синдром Меккеля |
AR |
76 |
91 |
|
CEP104
|
синдром Жубера |
AR |
7 |
5 |
|
CEP120
|
Короткореберная торакальная дисплазия 13 с полидактилией или без нее |
AR |
9 |
9 |
|
CEP164
|
Нефронофтиз |
AR |
11 |
9 |
|
CEP290
|
Синдром Барде-Бидля, врожденный амавроз Лебера, синдром Жубера, синдром Сениора-Локена, синдром Меккеля |
AR |
130 |
289 |
|
CEP41
|
синдром Жубера |
AR/Digenic |
7 |
11 |
|
CSPP1
|
Удушающая торакальная дистрофия Жена, синдром Жубера |
AR |
32 |
27 |
|
INPP5E
|
Синдром Жубера, умственная отсталость, ожирение туловища, дистрофия сетчатки и микропенис (синдром MORM) |
AR |
25 |
50 |
|
KIAA0556
|
Синдром Жубера 26 |
AR |
2 |
2 |
|
KIAA0586
|
Короткая торакальная дисплазия ребер с полидактилией, синдром Жубера |
AR |
29 |
31 |
|
KIAA0753
|
Орофациодигитальный синдром XV |
AR |
6 |
7 |
|
KIF7
|
Синдром акрокаллоза, синдром гидролеталуса, синдром Аль-Газали-Бакалиновой, синдром Жубера |
AR/Digenic |
24 |
44 |
|
MKS1
|
синдром Барде-Бидля, синдром Меккеля |
AR |
50 |
52 |
|
NPHP1
|
Нефронофтиз, синдром Жубера, синдром Сениора-Локена |
AR |
19 |
76 |
|
NPHP3
|
Нефронофтиз, почечно-печеночно-панкреатическая дисплазия, синдром Меккеля |
AR |
38 |
75 |
|
OFD1
|
Синдром Симпсона-Голаби-Бемеля, пигментный ретинит, орофациодигитальный синдром, синдром Жубера |
XL |
153 |
160 |
|
PDE6D
|
Синдром Жубера 22 |
AR |
3 |
1 |
|
RPGRIP1L
|
синдром COACH, синдром Жубера, синдром Меккеля, дегенерация сетчатки при цилиопатии, модификатор |
AR |
39 |
49 |
|
TCTN1
|
синдром Жубера |
AR |
6 |
6 |
|
TCTN2
|
Синдром Жубера, синдром Меккеля |
AR |
20 |
15 |
|
TCTN3
|
Орофациодигитальный синдром (синдром Мора-Маевского), синдром Жубера |
AR |
9 |
12 |
|
TMEM107
|
синдром Жубера |
AR |
10 |
3 |
|
TMEM138
|
синдром Жубера |
AR |
6 |
8 |
|
TMEM216
|
Синдром Жубера, синдром Меккеля |
AR |
17 |
8 |
|
TMEM231
|
Синдром Жубера, синдром Меккеля |
AR |
12 |
19 |
|
TMEM237
|
синдром Жубера |
AR |
7 |
11 |
|
TMEM67
|
Нефронофтиз, синдром COACH, синдром Жубера, синдром Меккеля |
AR |
87 |
170 |
|
TTC21B
|
Короткореберная торакальная дисплазия, нефронофтиз, асфиксическая торакальная дисплазия (ATD; Jeune) |
AR |
23 |
63 |
|
ZNF423
|
Нефронофтиз, синдром Жубера |
AD/AR |
10 |
7 |
* Тип наследования: аутосомно-доминантный (AD), аутосомно-рецессивный (AR), митохондриальный (mi), Х-сцепленный (XL), Х-сцепленный доминантный (XLD) и Х-сцепленный рецессивный (XLR);
** ClinVar, HGMD - количество вариантов гена, классифицированных как патогенные или вероятно патогенные в базе данных ClinVar, HGMD.
Возможная персонализация панели
В панель разрешается добавить дополнительные гены (до 200), которые связаны с симптомами, и удалить любые гены (чтобы осталось не меньше 2). Стоимость панели может меняться, а именно:
- малая панель 2-25 генов ,
- средняя панель 26-125 генов ,
- большая панель более 126 генов .
Преимущества этого теста
- Лаборатория (США), аккредитованная САР
- Персонал, сертифицированный CLIA, выполняющий клинические тесты в лаборатории, сертифицированной CLIA
- Мощные технологии секвенирования, передовые методы обогащения целей и точные биоинформатические пайплайны обеспечивают превосходную аналитическую производительность
- Тщательное составление клинически эффективных и научно обоснованных генных панелей
- Анализ некодирующих вариантов, вызывающих заболевания
- Анализ делеций и дупликаций (CNV)
- Строгая схема классификации вариантов
- Систематический рабочий процесс клинической интерпретации с использованием проприетарного программного обеспечения, обеспечивающего точную и прозрачную обработку данных NGS.
- Подробнейшее клиническое заключение на английском и украинском языках
- Возможность заказать сырые данные в формате .vcf
Что дальше?
Если анализ выявил варианты, являющиеся возможной причиной болезни, можно проверить наличие этих вариантов у родителей. Анализ отдельных мутаций по Сэнгеру.
Если анализ не выявил вариантов, являющиеся возможной причиной болезни, можно расширить тестирование до полного экзома Расширение панели до полного экзома. Кроме того, возможно получить сырые данные и делать повторную оценку каждые несколько лет. Возможно, со временем науке станут известны новые генетические факторы заболевания.
Как пройти исследование
Специальной подготовки не требуется: не нужно натощак, обильно пить воду.
Проводится забор венозной крови в пробирку с ЭДТА 4 мл.
Забор материала у нас в центре или можно передать нам пробирку ЭДТА, взятую в любой лаборатории. О возможности забора у детей уточнять дополнительно.
В наших пунктах забора, кроме Киева, забор материала стоит .
Необходимо предоставить заключение врача с клинической картиной (описание симтомов, диагноз), заполнить в электронном виде анкету.
Анализ не проводится для здоровых людей без сипмтомов, в этом случае рекомендуются скрининговые анализы на носительство.
По результатам секвенирования необходимо получить консультацию врача-генетика.
 Киев
Киев
