До панелі Синдром Жубера входить 36 генів, а також включена оцінка некодуючих варіантів, виявлення делецій і дублікатів.
Ідеально підходить для пацієнтів з клінічним подозрением на синдром Жубера.
Гени цієї панелі включені в панель Ціліопатії та панель Дистрофії сітківки.
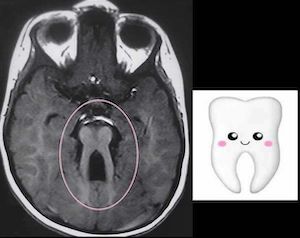 Класичний синдром Жубера (JS) характеризується тремя основними ознаками: відмінною аномалією розвитку мозкової залози та ствола мозку, так званим ознакою кореневого зуба (МТС), гіпотонією та затримкою розвитку. МТС, видимий на магнітно-резонансній томографії (МРТ), є результатом гіпоплазії черв'яка мозкового мозку та аномалії розвитку середнього і заднього мозку. Додаткові признаки, пов'язані з синдромом Жубера, включають аномалії сетчатки, ністагм, атаксію, полідактилію, фіброз печіння і захворювання почек. Оцінки поширення змінюються від 1:80 000 до 1:100 000, але це може бути заниженою оцінкою. У цілому близько 50% людей з синдромом Жубера і пов'язаними з ним розладами мають варіанти, ідентифіковані в одному з ідентифікованих генів. Дефектна біологія ресничек лежить в основі JS, і вона розділяє суттєвий аллелізм з іншими циліопатіями.
Класичний синдром Жубера (JS) характеризується тремя основними ознаками: відмінною аномалією розвитку мозкової залози та ствола мозку, так званим ознакою кореневого зуба (МТС), гіпотонією та затримкою розвитку. МТС, видимий на магнітно-резонансній томографії (МРТ), є результатом гіпоплазії черв'яка мозкового мозку та аномалії розвитку середнього і заднього мозку. Додаткові признаки, пов'язані з синдромом Жубера, включають аномалії сетчатки, ністагм, атаксію, полідактилію, фіброз печіння і захворювання почек. Оцінки поширення змінюються від 1:80 000 до 1:100 000, але це може бути заниженою оцінкою. У цілому близько 50% людей з синдромом Жубера і пов'язаними з ним розладами мають варіанти, ідентифіковані в одному з ідентифікованих генів. Дефектна біологія ресничек лежить в основі JS, і вона розділяє суттєвий аллелізм з іншими циліопатіями.
Гени, що входять до панелі:
| Ген |
Асоційований фенотип |
Спадкування* |
ClinVar** |
HGMD** |
|
AHI1
|
синдром Жубера |
AR |
62 |
93 |
|
ARL13B
|
синдром Жубера |
AR |
11 |
10 |
|
ARMC9
|
Синдром Жубера 30 |
AR |
12 |
11 |
|
B9D1
|
синдром Меккеля |
AR |
7 |
10 |
|
B9D2
|
синдром Меккеля |
AR |
8 |
4 |
|
C21ORF2
|
Дистрофія сітківки з макулярною стафіломою або без неї (RDMS), спондилометафізарна дисплазія, аксіальна (SMDAX) |
AR |
13 |
22 |
|
C5ORF42
|
Орофаціодигітальний синдром VI, синдром Жубера 17 |
AR |
97 |
103 |
|
CC2D2A
|
синдром COACH, синдром Жубера, синдром Меккеля |
AR |
76 |
91 |
|
CEP104
|
синдром Жубера |
AR |
7 |
5 |
|
CEP120
|
Короткоріберна торакальна дисплазія 13 з полідактилією або без неї |
AR |
9 |
9 |
|
CEP164
|
Нефронофтіз |
AR |
11 |
9 |
|
CEP290
|
Синдром Барде-Бідля, вроджений амавроз Лебера, синдром Жубера, синдром Сеніора-Локена, синдром Меккеля |
AR |
130 |
289 |
|
CEP41
|
синдром Жубера |
AR/Digenic |
7 |
11 |
|
CSPP1
|
Задушлива торакальна дистрофія Дружина, синдром Жубера |
AR |
32 |
27 |
|
INPP5E
|
Синдром Жубера, розумова відсталість, ожиріння тулуба, дистрофія сітківки та мікропеніс (синдром MORM) |
AR |
25 |
50 |
|
KIAA0556
|
Синдром Жубера 26 |
AR |
2 |
2 |
|
KIAA0586
|
Коротка торакальна дисплазія ребер з полідактилією, синдром Жубера |
AR |
29 |
31 |
|
KIAA0753
|
Орофаціодигітальний синдром XV |
AR |
6 |
7 |
|
KIF7
|
Синдром акрокаллозу, синдром гідролеталусу, синдром Аль-Газалі-Бакалінової, синдром Жубера |
AR/Digenic |
24 |
44 |
|
MKS1
|
синдром Барде-Бідля, синдром Меккеля |
AR |
50 |
52 |
|
NPHP1
|
Нефронофтіз, синдром Жубера, синдром Сеніора-Локена |
AR |
19 |
76 |
|
NPHP3
|
Нефронофтіз, нирково-печінково-панкреатична дисплазія, синдром Меккеля |
AR |
38 |
75 |
|
OFD1
|
Синдром Сімпсона-Голабі-Бемеля, пігментний ретиніт, орофаціодигітальний синдром, синдром Жубера |
XL |
153 | 160 |
|
PDE6D
|
Синдром Жубера 22 |
AR |
3 |
1 |
|
RPGRIP1L
|
синдром COACH, синдром Жубера, синдром Меккеля, дегенерація сітківки при циліопатії, модифікатор |
AR |
39 |
49 |
|
TCTN1
|
синдром Жубера |
AR |
6 |
6 |
|
TCTN2
|
Синдром Жубера, синдром Меккеля |
AR |
20 |
15 |
|
TCTN3
|
Орофаціодигітальний синдром (синдром Мора-Маєвського), синдром Жубера |
AR |
9 |
12 |
|
TMEM107
|
синдром Жубера |
AR |
10 |
3 |
|
TMEM138
|
синдром Жубера |
AR |
6 |
8 |
|
TMEM216
|
Синдром Жубера, синдром Меккеля |
AR |
17 |
8 |
|
TMEM231
|
Синдром Жубера, синдром Меккеля |
AR |
12 |
19 |
|
TMEM237
|
синдром Жубера |
AR |
7 |
11 |
|
TMEM67
|
Нефронофтіз, синдром COACH, синдром Жубера, синдром Меккеля |
AR |
87 |
170 |
|
TTC21B
|
Короткоберна торакальна дисплазія, нефронофтиз, асфіксічна торакальна дисплазія (ATD; Jeune) |
AR |
23 |
63 |
|
ZNF423
|
Нефронофтіз, синдром Жубера |
AD/AR |
10 |
7 |
* Тип успадкування: аутосомно-домінантний (AD), аутосомно-рецесивний (AR), мітохондріальний (mi), Х-зчеплений (XL), Х-зчеплений домінантний (XLD) та Х-зчеплений рецесивний (XLR);
** ClinVar, HGMD - кількість варіантів гена, класифікованих як патогенні або ймовірно патогенні в базі даних ClinVar, HGMD.
Можлива персоналізація панелі
В панель дозволяється додати додаткові гени (до 200), пов'язані з симптомами, та видалити будь-які гени (щоб залишилося не менше 2). Вартість панелі може змінюватися, а саме:
- мала панель 2-25 генів ,
- середня панель 26-125 генів ,
- велика панель понад 126 генів .
Переваги цього тесту
- Лабораторія (США), акредитована САР
- Персонал, сертифікований CLIA, проводить клінічні випробування у лабораторії, сертифікованій CLIA.
- Потужні технології секвенування, передові методи збагачення мішеней та біоінформатичні пайплайни забезпечують точну аналітичну продуктивність.
- Ретельне складання клінічно ефективних та науково обґрунтованих генних панелей
- Аналіз некодуючих варіантів, що викликають захворювання
- Строга схема класифікації варіантів
- Систематичний робочий процес клінічної інтерпретації з використанням пропрієтарного програмного забезпечення, що забезпечує точну та простежувану обробку даних NGS.
- Докладний клінічний висновок
- Можна замовити сирі дані секвенування в форматі.vcf
Що далі?
Якщо аналіз виявив варіанти, що є можливою причиною хвороби, можна перевірити наявність цих варіантів у батьків. Аналіз окремих мутацій по Сенгеру.
Якщо аналіз не виявив варіантів, які є можливою причиною хвороби, можна розширити тестування до повного екзома Розширення панелі до повного екзома. Крім того, можна отримати сирі дані і робити повторну оцінку кожні кілька років. Можливо, згодом науці стануть відомі нові генетичні чинники захворювання.
Як пройти дослідження
Натще не потрібно, рясно пити воду.
Проводиться забір венозної крові в пробірку з ЕДТА 4 мл.
Забір матеріалу у нас у центрі або можна передати нам пробірку ЕДТА, взяту у будь-якій лабораторії. Про можливість забору у дітей уточнювати додатково.
У наших пунктах забору крім Києва забор матеріалу коштує .
Необхідно надати висновок лікаря з клінічною картиною (опис симтомів, діагноз), заповнити в електронному вигляді анкету.
Аналіз не проводиться для здорових людей без симптомів, у цьому випадку рекомендуються скринінгові аналізи на носійство.
За результатами секвенування необхідно отримати консультацію лікаря-генетика.
 Київ
Київ
