


Синдром Клайнфельтера (XXY)
Дата: 15.06.2016
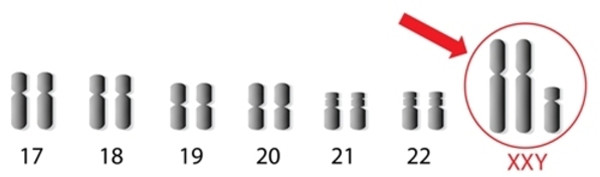
Синдром Клайнфельтера - патологія, яка характеризується наявністю у хлопчиків зайвої X хромосоми (мінімум однієї), внаслідок чого порушується їхнє статеве дозрівання.
Синдром Клайнфельтера – патологія, яка характеризується наявністю у хлопчиків зайвої X хромосоми (мінімум однієї), внаслідок чого порушується їхнє статеве дозрівання. Захворювання 1942 року вперше було описано Клайнфельтером.
Хвороба Клайнфельтера – одна з найпоширеніших хромосомних аномалій, що зустрічається частіше за синдром Дауна. Поширеність захворювання приблизно 1:500. Діагностувати цю патологію в неонатальний період неможливо, так у хлопчиків відразу після народження немає жодних характерних ознак.
![]() Дізнатися з точністю 99% про ризик синдрому Клайнфельтера та інших хромосомних аномалій, а також підлогу плода, можна з 9 тижнів вагітності, всього лише здавши кров із вени. Докладніше про НІПТ-тест.
Дізнатися з точністю 99% про ризик синдрому Клайнфельтера та інших хромосомних аномалій, а також підлогу плода, можна з 9 тижнів вагітності, всього лише здавши кров із вени. Докладніше про НІПТ-тест.
Ознаки Синдрому Клайнфельтера
Синдром Клайнфельтера можна запідозрити до підліткового віку за такими ознаками:
- щодо зростання, довгі ноги, високе розташування талії;
- у деяких хворих відставання від однолітків у розумовому розвитку;
- емоційна лабільність (чергування радості та смутку, зміна настрою з незначного приводу);
- наявність вроджених вад розвитку (серця та інших органів), які виявляються при плановому обстеженні і можуть супроводжувати спадковим порушенням.
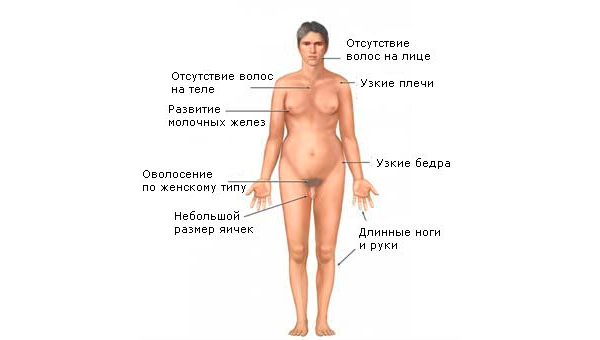
У підлітковому віці з'являються чітко виражені фізичні та психічні ознаки захворювання. Надалі такі хлопчики стають безплідними, в їхній насінній рідині немає життєздатних сперматозоїдів.
Можливі ускладнення
Затримка розумового розвитку (іноді до дебільності);психічні порушення, що згодом визначають схильність до алкоголізму, суїциду, асоціальної поведінки;безплідність, проте в даний час можливе запліднення в пробірці (екстракорпоральне запліднення), але дана методика у цих хворих ще продовжує та широко не застосовується; на тлі ожиріння високий ризик розвитку цукрового діабету.
Ускладнення захворювання та їх тяжкість залежить від термінів початку лікування (що раніше поставлено діагноз і розпочато гормональну терапію, тим менше ускладнень).
Прогноз
Прогноз для життя є сприятливим. При своєчасній діагностиці та лікуванні захворювання ризик розвитку ускладнень мінімальний, хворі добре адаптовані до життя. Останніми роками активно вирішується проблема безпліддя таких пацієнтів.
Пренатальна діагностика
1) Інвазивні дослідження (амніоцентез, біопсія хоріону) в основному призначають тим жінкам, у яких спостерігається підвищений ризик того, що народиться малюка із синдромом Едвардса, наприклад, пацієнткам, чий вік перевищує 35 років або з поганими результатами неінвазивних тестів: УЗД та аналізів. Інвазивні методи діагностики є високоточними, проте, враховуючи ризик ускладнень, не підходять для масового проведення всім вагітним, а проводяться лише за особливими показаннями.
2) Неінвазивні технології, так звані скринінги. Скринінг – комплексне дослідження вагітних жінок на наявність у плода хромосомних аномалій. Виділено кілька ознак, що вказують на високий ризик наявності захворювання, які можуть виявити УЗД плода (відсутність носової кістки, збільшена товщина комірного простору, недостатня довжина стегнових та плечових кісток та інші особливості). У комплексі з УЗД йде біохімічний аналіз крові матері на такі гормони як вільний бета-ХГЛ та PAPP-A. Отримані дані з біохімічних маркерів аналізують у сукупності з результатами ультразвукового дослідження, а результат всього скринінгу є розрахунком ризику наявності хромосомної аномалії у плода.
Однак при використанні стандартних тестів на синдром Едвардса, лише у 3% жінок, спрямованих на інвазивну діагностику, дійсно підтверджується наявність захворювання. У той же час не виключені й хибно-негативні результати, коли скринінг показує низький ризик, а дитина народжується із хромосомною патологією.
- точність 99%, що набагато точніше за класичну діагностику (УЗД та біохімічний скринінг)
- цілком безпечний, на відміну від інвазивних методик - для забору матеріалу на аналіз необхідно просто взяти кров із вени вагітної жінки.
- на ранніх термінах: аналіз можна проводити вже на 9-му тижні вагітності.

Генетик